I want to announce that on 19th October I’ll teach the workshop titled “Introduction to Bioinformatics applied to Metagenomics and Community Ecology” during the conference Community Ecology for the 21st Century (Évora, Portugal).
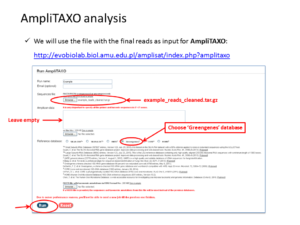
In this workshop I’ll introduce the new tool called AmpliTAXO that allows an online, easy and automated analysis of NGS data from ribosomal RNA and other genetic markers used in metagenomics.
If you are interested, you can contact here with the conference organizers to join the workshop or the full conference, there are still available places in the workshop.
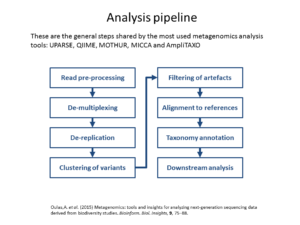
The workshop will consist in two modules, in the first, will be exposed the metagenomics fundamentals, challenges, the technical advances of the high-throughput sequencing techniques and the analysis pipeline of the most used tools (UPARSE, QIIME, MOTHUR). The second part will be practical and we will perform an analysis of real metagenomic data from NGS experiments.
Below you can read the abstract an a small advance of the contents…

Abstract
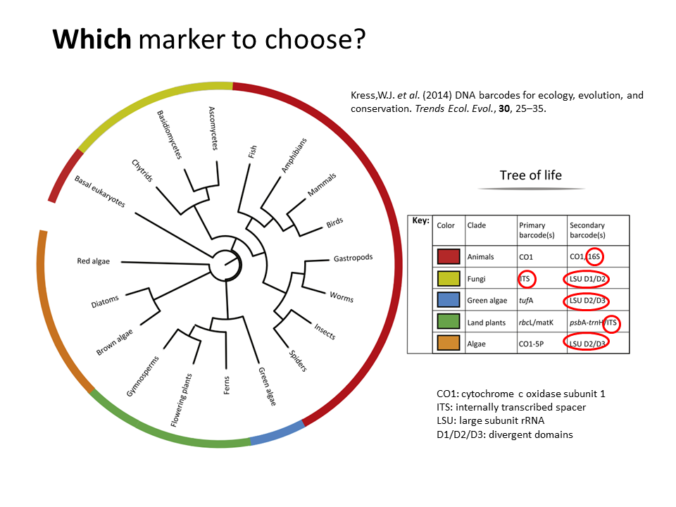
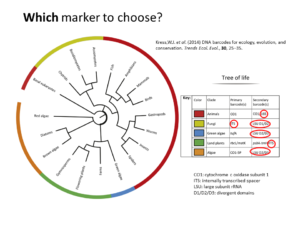
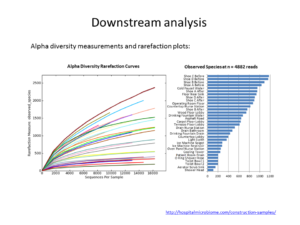
Metagenomics (also referred to as ‘environmental’ or ‘community’ genomics) is the study of genetic material recovered directly from environmental samples. This discipline applies a suite of genomic technologies and bioinformatics tools to directly access the genetic content of entire communities of organisms. There has been a metagenomics revolution over the past 5-10 years, with substantial contributions to the field of microbial community ecology. This success has derived from a number of techniques and protocols that are impossible to cover in a single course. In the present workshop, the major Next Generation Sequencing approaches in metagenomics will be introduced. We will focus on the Amplicon Sequencing (AS) technique that allows us to identify thousands of species from databases in dozens of samples with a single experiment. AS mainly consists of parallel PCR amplification of DNA from biological markers (e.g. 16S rRNA gene) that serve as universal DNA barcodes, to identify with great accuracy species from across the Tree of Life. We will go through all the steps of the Bioinformatics analysis pipeline: from read merging, de-multiplexing, de-replication, clustering, error correction and chimera removal to alignment of OTU representative variants against reference databases and taxonomical annotation. Each analysis stage will be explained with examples and a real data analysis will be performed with AmpliTAXO. AmpiTAXO is an easy-to-use online server for one-step metagenomics analysis, in contrast to other popular multi-step analysis tools like UPARSE, QIIME or MOTHUR.
Leave a Reply