
 BLAST changed some time ago for better and faster to BLAST+ version, but along the way small differences were introduced that may confound more than one.
BLAST changed some time ago for better and faster to BLAST+ version, but along the way small differences were introduced that may confound more than one.
Old BLAST was run with the command ‘blastall‘. In this way, a protein alignment should be called as ‘blastall -p blastp‘ and the same for nucleotides should be ‘blastall -p blastn‘, and if we want to use MEGABLAST then it exists the different command ‘megablast‘.
Nevertheless, in the ‘new’ BLAST+ version, the alignments of proteins and nucleotides were splitted in two different commands: ‘blastp‘ and ‘blastn‘ (see manual).
Till here, everything looks natural and logic, but not everyone knows that THE DEFAULT OPTION IN THE NEW BLASTN COMMAND IS MEGABLAST.
If we run ‘blastn -help‘ we will obtain the following explanation:
*** General search options -task <String, Permissible values: 'blastn' 'blastn-short' 'dc-megablast' 'megablast' 'rmblastn' > Task to execute Default = `megablast'
But not everyone is interested in increasing the alignment speed, because many of us that still use Blastn is because we appreciate its great sensitivity to detect short local alignments. Nowadays Blast can align thousands of sequences in a reasonable time of minutes or even seconds. To increase the alignment speed in alignments with millions of sequences involved there are other better alternatives as Bowtie2.
As CONCLUSION: if we use Blastn and we are interested in a high sensitivity, we should run it as:
blastn -task blastn
If not, we will execute MEGABLAST and we can lose a great sensitivity and risk to not to find the expected alignments.
As example, let’s search homology between the human 2’beta microglobuline (NM_004048.2) and murine one (NM_009735.3) using online version of Blastn with default options (‘Highly similar sequences – megablast’):
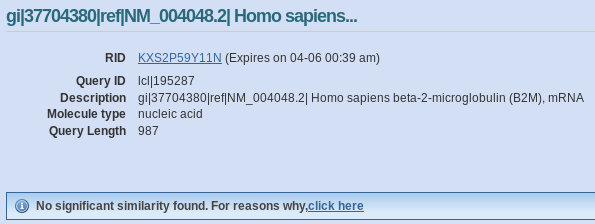
But if we change the search option to ‘Somewhat similar sequences (blastn)’:

It is a big difference, megablast does not find any significant similarity, whereas blastn outputs a high similarity alignment with an E-value of 1.5E-56!!!!

