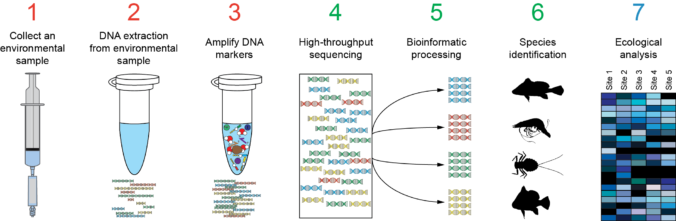
In the previous post I explained the fundamentals about the Amplicon Sequencing (AS) technique, today I will show some current and future applications in HLA-typing.
Other field of use of AS is the genotyping of complex gene families. For example, the major histocompatibility complex (MHC). This gene family is known to be highly polymorphic (high allele variation) and to have multiple copies of an ancestor gene (paralogues). MHC genes of class I and II codify the cellular receptors that present antigens to immune cells. MHC in humans is also called human leukocyte antigen (HLA). HLA-typing has a key role in the compatibility upon any tissue transplantation and has been associated with more than 100 different diseases (primarily autoimmune diseases) and recently is associated to various drug positive and negative responses. HLA loci are so polymorphic that there are not 2 individuals in a non-endogamic population with the same set of alleles (except twins).
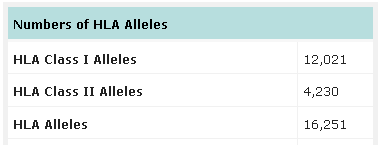
Number of HLA alleles known up to date. Source: IMGT-HLA database
As in personalized medicine and metagenomics/metabarcoding, there are 2 approaches for NGS HLA-typing: the first is to use the whole genomic, exomic or transcriptome data and the second is to amplify specific HLA loci regions by amplicon sequencing. Second approach is suitable for typing hundreds/thousands of individuals but requires tested primers for multiplex PCR of HLA regions.
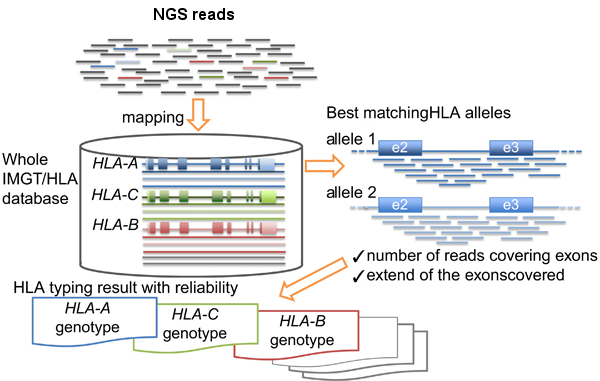
Basically the HLA-typing analysis workflow after sequencing the PCR products, consists in:
- Map/align the reads against HLA allele reference sequences from the IMGT-HLA public database.
- Retrieve the genotypes from the references with longer and better mapping scores.
Inoue et al. wrote a complete review about the topic in ‘The impact of next-generation sequencing technologies on HLA research‘.
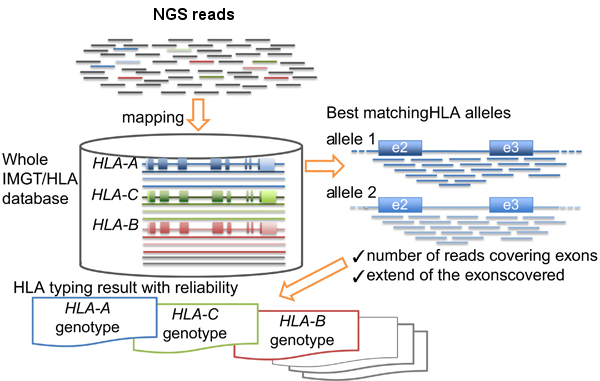
HLA-typing workflow. Modified from Inoue et al.
Nowadays there are commercial kits that allow reliable, fast and economic HLA-typing: Illumina TruSight HLA v2, Omixon Holotype HLA, GenDx NGSgo or One Lambda NXType NGS.