The story of this post started few months ago at a immunotherapies conference in Barcelona, at this time I was looking for a job in cancer research. I had a great time learning about new cancer therapies and meeting nice people, but few doctors were interested, or had money, to contract a bioinformatician. Nevertheless, one of them told me: “I don’t know you, but if you show me that you are able to discover tumor neoantigens, then I’ll consider to contract you in the future”. I hope this person will read the post 😉
But before starting, I want to dedicate this post to all the doctors, nurses, scientists and patients that fight everyday against cancer, specially those from Hospital Miguel Servet de Zaragoza and Clínica Universidad de Navarra that treat my father, also to many other brilliant and devoted people that I have met from places like Vall d’Hebron Hospital, Quiron Dexeus or 12 de Octubre, and finally, to my new team at Agenus Pharmaceuticals.
State of the art of tumor neoantigen discovery
With next generation sequencing (NGS) we are able to sequence the full genome of a tumor or at least of the coding regions or exome. If we identify with the help of NGS data which tumor-specific genes express mutant peptides that are able to activate T cells, then we can design vaccines to boost the natural immune response to the tumor, this mutant peptides are also called neoantigens. There is a nice and recent Nature Review about neoantigens discovery, and also there are interesting articles with practical cases of neoantigens discovery and personalize treatments for cancer patients (e.g. Robbins et al., Pritchard et al.).
But we are interested in the bioinformatics point of view, so let’s make a summary of some neoantigens discovery protocols and software in the literature:
- pVAC-Seq v4.0: a pipeline for the identification of personalized Variant Antigens by Cancer Sequencing (pVAC-Seq) that integrates tumor mutation and expression data (DNA- and RNA-Seq) (original article).
- TSNAD: an integrated software for cancer somatic mutation and tumour-specific neoantigen detection (original article).
- neoantigenR: an R package that identifies peptide epitope candidates (biorxir article).
- MuPeXI: mutant peptide extractor and informer, a tool for predicting neo-
epitopes from tumor sequencing data (original article). - INTEGRATE-neo: a pipeline for personalized gene fusion neoantigen discovery (original article).
We can conclude that the topic is quite new, most of the previous pipelines have been published during the last year, and that the neoantigen identification is a very complex task as we can see for example in the pVAC-Seq workflow schema:
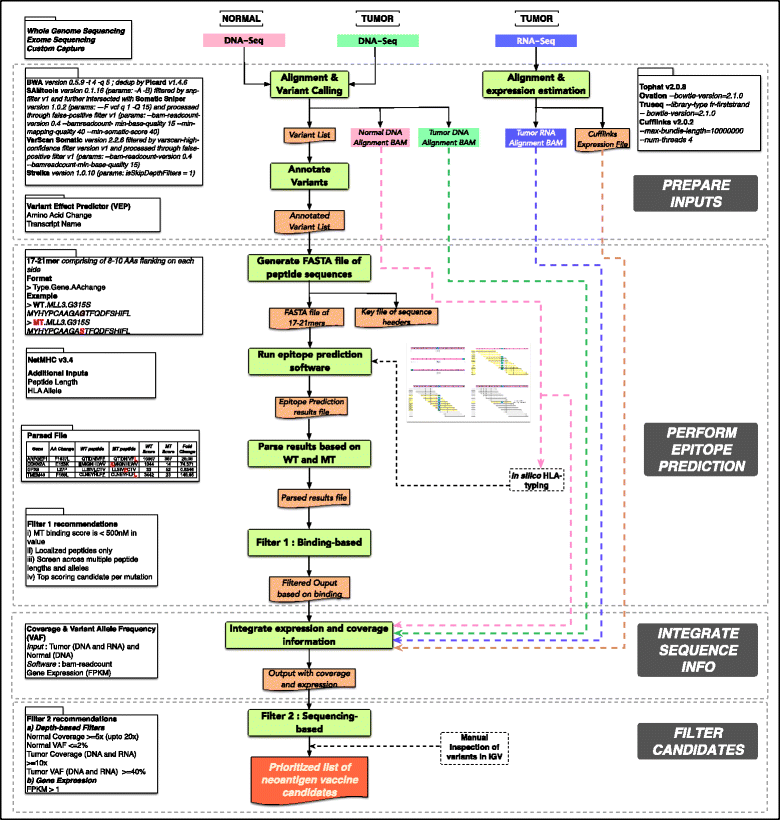
Overview of the pipeline pVAC-Seq
To show an example of neoantigens discovery pipeline, I’ll try to simplify the process and follow the Best Practices guide from The Broad Institute for somatic variants discovery:
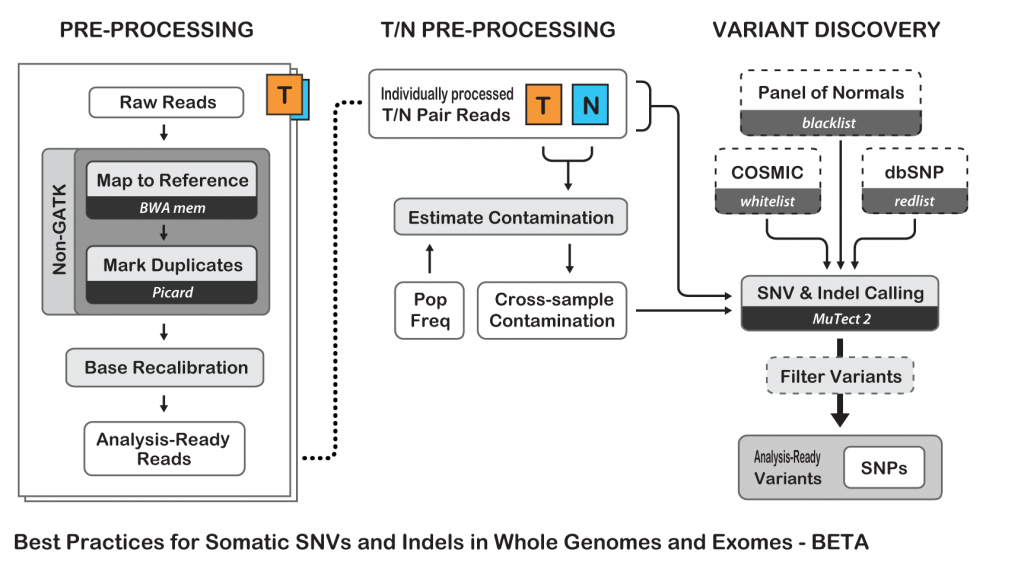
Best Practices for Somatic SNV and Indel Discovery
Tumor neoantigen discovery pipeline description
This protocol has been developed for didactic purposes and it is not formally validated neither intended for human diagnosis.
Basically I propose here a somatic variant calling pipeline with some additional filtering of the variants at the end. I tested the pipeline with whole exome sequencing (WES) Illumina paired-end reads from normal and tumor tissues.
The full pipeline can be summarized in 10 steps, here is briefly explanation of every step and some additional considerations about the chosen tools:
- Normal and tumor reads mapping, sorting and indexing:
- In the protocol I have chosen BWA-MEM as recommended in Cornish and Guda paper, original authors used Novoalign, but other aligners could be considered. In this aspect, aligners as Bowtie2 may be too sensitive and could report some extra false positive variants, but it’s just a personal opinion.
- Removal of duplicated reads:
- Maybe this step could be skipped as there are not many redundant reads and it’s unlikely that they map in variant positions. MarkDuplicates from Picard tools was used, I would suggest in the future to try ‘MarkDuplicatesWithMateCigar’ from the same suite, but I have not enough experience to recommend it.
- Adding sample/group information to the mapped reads:
- This is just a formal requirement for the variant callers to work properly and distinguish between normal and tumor groups of reads.
- Realignment around indels:
- This step is already integrated in the Mutect2 somatic variant caller and could be redundant, but other variant callers do not integrate it so it is strictly recommended to avoid false calling of indels and surrounding SNPs.
- Somatic variant calling with MuTect2:
- MuTect2 is integrated in the GATK suite, the basic operation of MuTect2 proceeds similarly to that of the popular HaplotypeCaller, with a few key differences, taking as input tumor and normal alignments and producing somatic variant calls only.
- Filtering of somatic variants:
- Some strict filters are imposed to discover only true somatic variants: a minimum depth of 10 reads and a variant frequency of at least 10%. Additionally, variants must be in coding regions of genes (it is filtered in the last step).
- Annotation of filtered somatic variants
- Filtered variants are annotated: gene and transcript names and IDs, chromosome position, nucleotide and protein mutations and other relevant information about the antigens is annotated in a tabulated, CSV or HTML human readable format
- Neoantigen peptides design
- Neoantigen peptides are designed extracting the amino acid sequence containing the variant (10-15 residues)
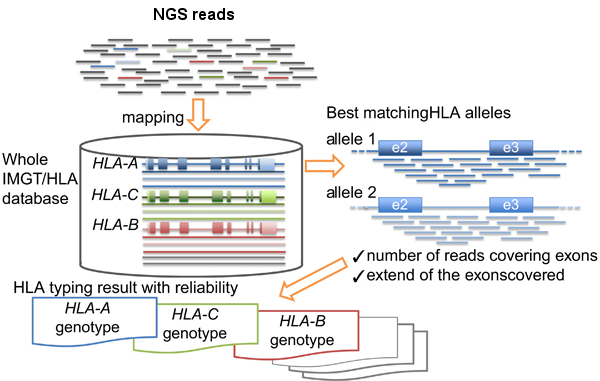
- HLA-typing with exome or RNA-seq data
- Normal and tumor or RNA-seq (more accurate) data will be used as input of AmpliHLA to retrieve the HLA alleles of the patient.
- HLA-binding affinity calculation and peptide ranking
- NetMHCpan software will calculate the binding affinities of the designed peptides to HLA alleles, these predictions together with expression data will be used to estimate the potential immunogenicity of the peptides.
Required tools and files
Before starting, you should install in your server or computer the following tools:
- BWA aligner
- SAMtools
- VCFtools
- Picard
- GATK 3.8 (GATK4 is still not functional)
- Variant filtering scripts
- AmpliHLA
- NetMHCpan
And download human genome files and SNP/indels annotations (be always consistent with the genome version and have in mind that new versions could be available):
- Human genome in FASTA format (e.g. Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz)
- Human curated indel database (e.g. Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz)
- The Single Nucleotide Polymorphism database (dbSNP, e.g. All_20170710.vcf.gz)
- The Catalogue Of Somatic Mutations In Cancer (COSMIC, e.g. CosmicCodingMuts.vcf.gz)
Previous files (or equivalent ones) can be also downloaded from Google Cloud repository (not tested).
After this long introduction, let’s start the analysis…
Somatic variant calling and tumor neoantigen discovery pipeline
To simplify I’ll consider that all the input files, including the genome and reads ones, are in the working directory and all the output files will be stored into the ‘output’ folder. All the comments are marked with ‘#’ to differentiate them from the commands. Programs will be installed in the standard ‘/opt’ folder in the Linux system and run with 30 threads when it is possible (number of parallel threads/processes should be adjusted depending of your server or computer). Analysis time will depend of the system and the number of parallel processes, in a small server with 30 threads it may take around 15-20 hours to be completed. Steps 8 and 10 are sill not fully automatized.
1. Normal and tumor reads mapping, sorting and indexing
# Index genome FASTA file for BWA mapping: /opt/bwa-0.7.15/bwa index Homo_sapiens.GRCh38.dna.primary_assembly.fa # Normal reads mapping, sorting and indexing with BWA-MEM: /opt/bwa-0.7.15/bwa mem -M -v 1 -t 30 Homo_sapiens.GRCh38.dna.primary_assembly.fa NORMAL_R1.fastq.gz NORMAL_R2.fastq.gz | /opt/samtools-1.6/bin/samtools view -Sb -F 4 -@ 30 | /opt/samtools-1.6/bin/samtools sort -@ 30 > output/NORMAL.bam ; /opt/samtools-1.6/bin/samtools index -@ 30 output/NORMAL.bam output/NORMAL.bai # Tumor reads mapping, sorting and indexing: /opt/bwa-0.7.15/bwa mem -M -v 1 -t 30 Homo_sapiens.GRCh38.dna.primary_assembly.fa TUMOR_R1.fastq.gz TUMOR_R2.fastq.gz | /opt/samtools-1.6/bin/samtools view -Sb -F 4 -@ 30 | /opt/samtools-1.6/bin/samtools sort -@ 30 > output/TUMOR.bam ; /opt/samtools-1.6/bin/samtools index -@ 30 output/TUMOR.bam output/TUMOR.bai
2. Removal of duplicated reads
# Removes normal duplicated reads: java -jar /opt/picard-2.14.0/build/libs/picard.jar MarkDuplicates VERBOSITY=ERROR VALIDATION_STRINGENCY=SILENT REMOVE_DUPLICATES=true I=output/NORMAL.bam O=output/NORMAL.dups_removed.bam M=output/NORMAL.metrics.txt # Removes tumor duplicated reads: java -jar /opt/picard-2.14.0/build/libs/picard.jar MarkDuplicates VERBOSITY=ERROR VALIDATION_STRINGENCY=SILENT REMOVE_DUPLICATES=true I=output/TUMOR.bam O=output/TUMOR.dups_removed.bam M=output/TUMOR.metrics.txt
3. Adding sample/group information to the mapped reads
# Adds normal read group, the name of the individual and the phenotype (3998_normal): java -jar /opt/picard-2.14.0/build/libs/picard.jar AddOrReplaceReadGroups VERBOSITY=ERROR VALIDATION_STRINGENCY=SILENT CREATE_INDEX=true SO=coordinate RGID=3998_normal RGLB=3998_normal RGPL=illumina RGPU=3998_normal RGSM=3998_normal I=output/NORMAL.dups_removed.bam O=output/NORMAL.grouped.bam # Adds tumor read group, the name of the individual and the phenotype (3998_tumor): java -jar /opt/picard-2.14.0/build/libs/picard.jar AddOrReplaceReadGroups VERBOSITY=ERROR VALIDATION_STRINGENCY=SILENT CREATE_INDEX=true SO=coordinate RGID=3998_tumor RGLB=3998_tumor RGPL=illumina RGPU=3998_tumor RGSM=3998_tumor I=output/TUMOR.dups_removed.bam O=output/TUMOR.grouped.bam
4. Realignment around indels
# Normal reads realignment around indels using the GATK modules RealignerTargetCreator and IndelRealigner: java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T RealignerTargetCreator -nt 30 -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I output/NORMAL.grouped.bam -known /Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz -o output/NORMAL.intervals ; java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T IndelRealigner -maxReads 100000 -maxInMemory 1000000 -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -targetIntervals output/NORMAL.intervals -I output/NORMAL.grouped.bam -known /Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz -o output/NORMAL.realign.bam # Tumor reads realignment around indels using the GATK modules RealignerTargetCreator and IndelRealigner: java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T RealignerTargetCreator -nt 30 -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I output/TUMOR.grouped.bam -known /Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz -o output/TUMOR.intervals ; java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T IndelRealigner -maxReads 100000 -maxInMemory 1000000 -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -targetIntervals output/TUMOR.intervals -I output/TUMOR.grouped.bam -known /Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz -o output/TUMOR.realign.bam
5. Somatic variant calling with Mutect2 (GATK tool)
# Creates dbSNP VCF index file required by GATK: gunzip /All_20170710.vcf.gz ; bgzip /All_20170710.vcf ; tabix -p vcf /All_20170710.vcf.gz # Base quality score recalibration for normal reads alignments (BQSR): java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T BaseRecalibrator -nct 30 -knownSites /All_20170710.vcf.gz -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I output/NORMAL.realign.bam -o output/NORMAL.bqsr ; java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T PrintReads -nct 30 -BQSR output/NORMAL.bqsr -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I output/NORMAL.realign.bam -o output/NORMAL.recalib.bam # Base quality score recalibration for tumor reads alignments (BQSR): java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T BaseRecalibrator -nct 30 -knownSites /All_20170710.vcf.gz -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I output/TUMOR.realign.bam -o output/TUMOR.bqsr ; java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T PrintReads -nct 30 -BQSR output/TUMOR.bqsr -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I output/TUMOR.realign.bam -o output/TUMOR.recalib.bam # Calls somatic variants (SNPs and indels) via local re-assembly of haplotypes with MuTect2: java -jar /opt/gatk-3.8/GenomeAnalysisTK.jar -T MuTect2 -nct 30 -D /All_20170710.vcf.gz -cosmic /CosmicCodingMuts.vcf.gz -R Homo_sapiens.GRCh38.dna.primary_assembly.fa -I:normal output/NORMAL.recalib.bam -I:tumor output/TUMOR.recalib.bam -o output/normal_vs_tumor.mutect2.all.vcf
6. Filtering of somatic variants
MuTect2 may retrieve more than 10000 variants, but after filtering the number is reduced around 8-10 times.
# Filters somatic variants called by each method: filter_somatic_variants.pl -pass -vardepth 10 -varfreq 0.1 -i output/normal_vs_tumor.mutect2.all.vcf -o output/normal_vs_tumor.mutect2.somatic.vcf
7. Annotation of filtered somatic variants
If we obtained around 1000 variants after previous filtering step, the annotation of only variants present in coding regions will reduce the number to few hundreds.
# Annotates final variants and prints the details in a tabulated format file: annotate_variants.pl -i output/normal_vs_tumor.mutect2.somatic.vcf > output/normal_vs_tumor.mutect2.somatic.txt
8. Neoantigen peptides design
# Creates a FASTA file with the sequences of the peptides (13 aa) containing the somatic mutations previously detected variants_to_peptides.pl -l 13 -i output/normal_vs_tumor.mutect2.somatic.vcf -o output/normal_vs_tumor.mutect2.peptides.fa
9. HLA-typing with exome or RNA-seq data
# Analyzes WES, or RNA-seq if available, data and retrieves the HLA alleles of the patient that will be used in the last step to calculate the affinities of the peptides. ampliHLA.pl -thr 30 -ty mapping -r gen -i NORMAL_R1.fastq.gz NORMAL_R2.fastq.gz > amplihla_normal.txt ampliHLA.pl -thr 30 -ty mapping -r gen -i TUMOR_R1.fastq.gz TUMOR_R2.fastq.gz > amplihla_tumor.txt
10. HLA-binding affinity calculation and peptide rankin
# Calculates and ranks the MHC-binding affinity netMHCpan -BA -xls -f output/normal_vs_tumor.mutect2.peptides.fa -a HLA-A01:01,HLA-A02:01,HLA-B15:01...
Finally I didn’t get any job in Spain, but I learned a lot 🙂